The baby's development is delayed.
Author:Lilac mother Time:2022.07.28

A child is enough to change a family.
The 5 -year -old Xiaodong (pseudonym) face is no different from other children, but unlike other children, Xiao Dong cannot stand on and walk.
In fact, parents have taken a number of hospitals with Xiao Dong, from onset to confirmation, and used for 4 years.
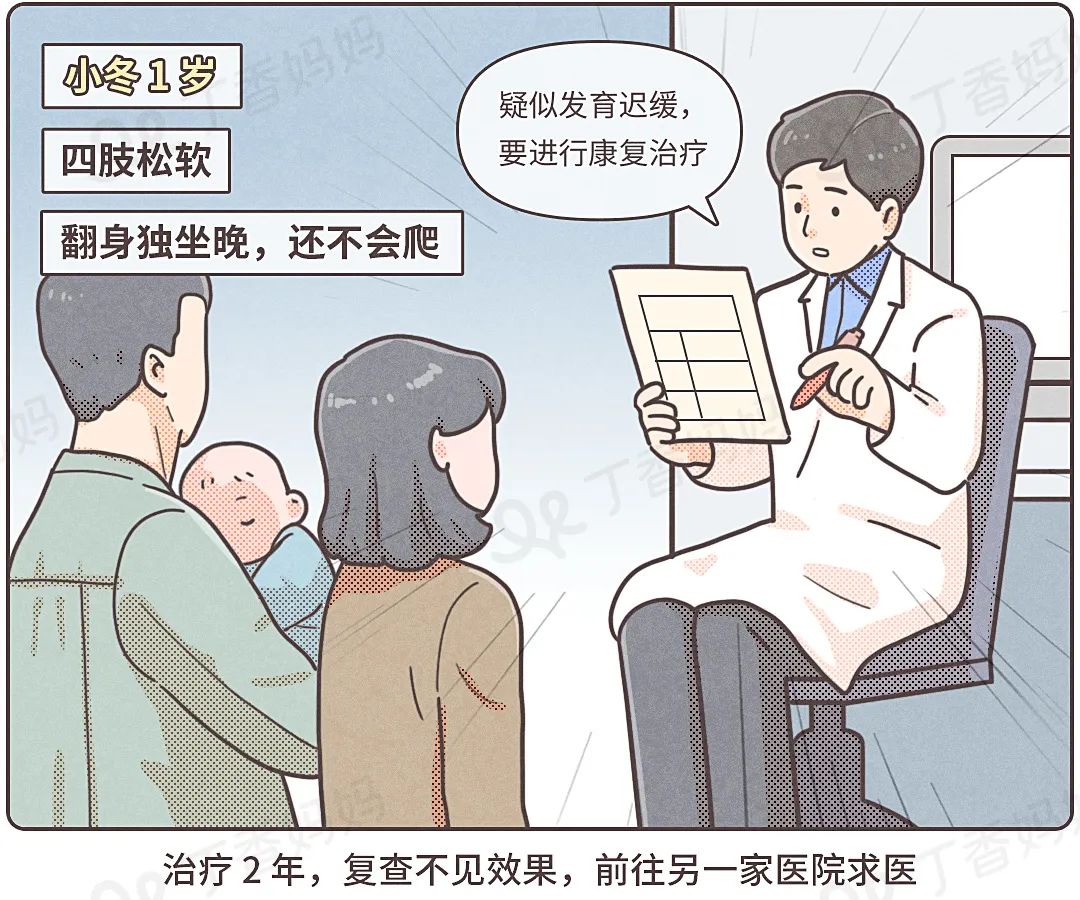

Slide left to check the experience of Xiao Dong for a doctor
In the past four years, her mother resigned from work and took care of Xiaodong full -time; Dad was responsible for making money and raising Xiao Dong's medical expenses.
From suspected delayed development, severe muscle weakness to the final diagnosis, 4 years of sadness.
If I have a chance, I really hope that I will get sick, if I can confirm and treat it early, Xiao Dong may be able to leave now
This is the voice of Xiao Dong's mother, and the voice of parents of countless rare sick families.
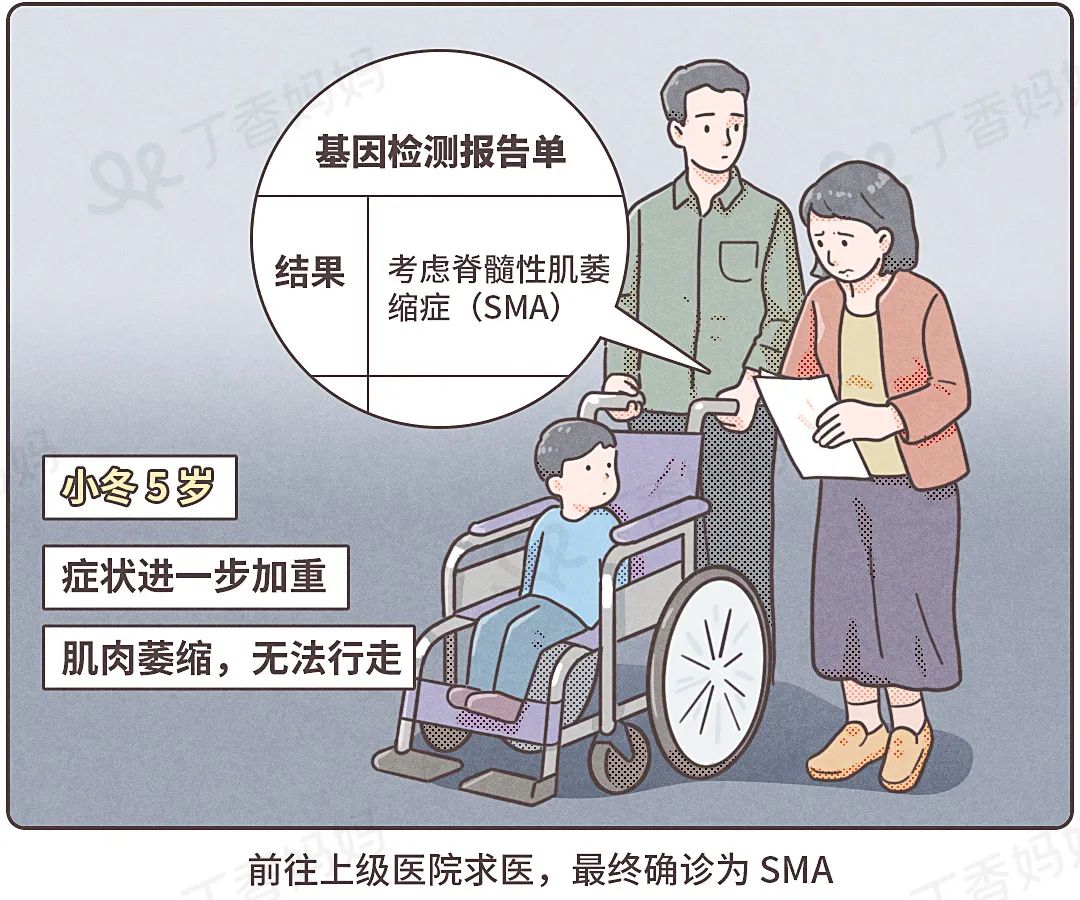
That's right, Xiao Dong's final diagnosis is an extremely serious rare genetic disease -spinal muscle atrophy (SMA).
SMN1 gene mutation
The culprit of muscle loss of strength
SMA is a recessive inherited genetic disease [1], which will cause the muscles of the fetus, infants and even adults to lose strength and gradually shrink. The cause is pathogenic variation of motor neuron survival gene 1 (SMN1).
Acturavity, that is, there is no gender bias, regardless of whether men and women will be affected; hiddenness means that only when the genes of both parents carry abnormal genes, the next generation may be sick.
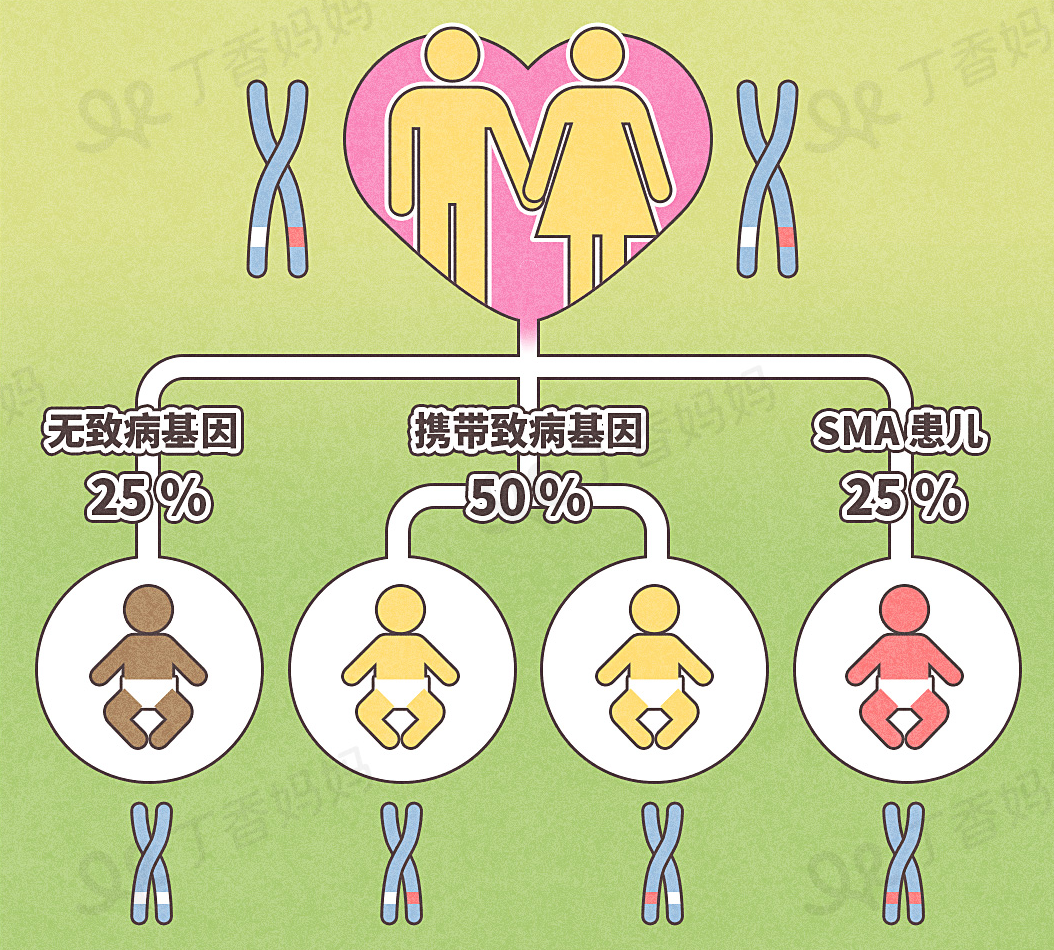
Picture: Parents carrying mutant genes with a 3/4 chance to inherit the mutant gene to the next generation.
Although spinal muscle atrophy is divided into rare diseases, this disease is ranked first among all deadly genetic diseases under the age of 2 [1,2].
Domestic studies have shown that of about every 42 people in the Chinese population, one person carries defective genes [3]. Among the surviving newborns, about every 10,000 are diagnosed [3].
Why does this genetic defects gradually lose their muscles?
Human genes are like a recipe, each of which is the duty of various genes to continue to make key nutrients for the human body -protein with different functions.
As one of the recipes, the protein produced by the SMN1 gene is called SMN [4]. This protein is very important for motor neurons connecting muscles.
Babies carrying two mutant SMN1 genes will be tampered with the steps of making SMN proteins. As a result, they can only get a variable -flavored, completely non -effect protein, making the above neurons survive [5]. The muscles have no nerve stimulation, and they can't force it, and they gradually shrink.
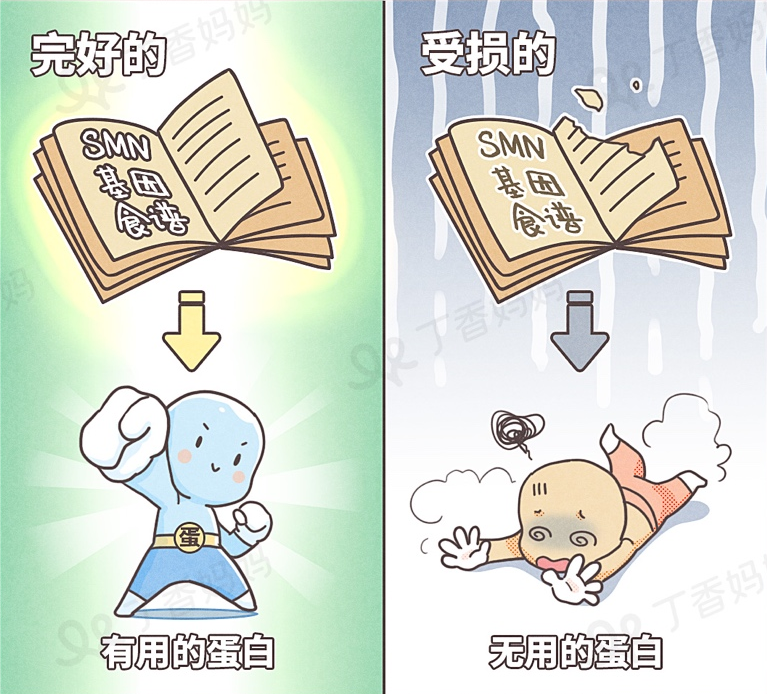
However, not all infants and young children are the same. In addition to the SMN1 gene, there is a spare SMN2 gene in humans [6], but the number of SMN2 in different patients is different.
SMN2 is like another recipe for making SMN proteins but inaccurate steps. In most cases, it can only make SMN proteins that have changed. When luck, you can get a normal SMN protein accidentally. SMA patients can only rely on these very limited normal SMN proteins to maintain the function of motor neurons.
Obviously, if the two SMN1 genes in the child have changed, the severity depends on the number and luck of the SMN2 gene. This explains why some people slowly begin to show a state of disease until they are adults.
Same disease
The baby's life may be very different
Five -year -old Xiao Dong, from the four years of onset to the diagnosis, because the speed of normal SMN protein in the body is increasingly unable to keep up with physical needs, and SMN protein is closely related to the survival of the motor nerve to control muscles, so I lost my legs. The neurons can no longer stand and walk.
In this way, can this situation be reversed before these motor neurons have been treated before apopping?
The answer is yes.
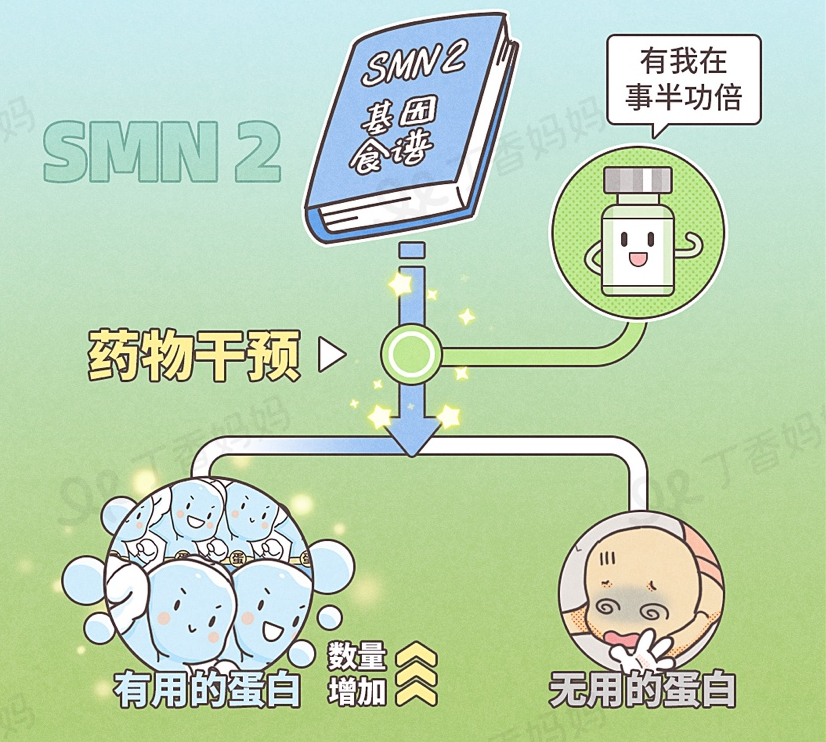
Remember the SMN2 gene mentioned earlier? Since the child's SMN1 gene is no longer reliable, various drugs are basically revised this disease around the genes of SMN2 and its derivatives.
Because the SMN2 gene can occasionally make normal SMN proteins, the drug only needs to increase the success rate of normal protein, and neurons may be saved [7-8].
In March 2021, the Department of Neurology, Shenzhen Children's Hospital moved to a baby Xiaohong (a pseudonym) with a SMN1 mutant. Mother is a SMA patient who is 3 years old, traveling to a wheelchair; dad is likely to be a mutant gene. Carrier [9].
If you do not intervene, according to the natural course of the disease, only 1 month of Xiaohong will likely cause disease within half a year old. The muscles controlled by the breathing before the age of two will be affected and eventually endanger life due to respiratory failure.
Because her mother was also a patient with SMA, Xiaohong performed genetic testing early after birth. After a month, SMA symptoms were admitted to the hospital for correction. As a result, the outpatient clinic of October found that Xiaohong's mileage milestone was similar to normal children of the same age!
Unfortunately, many children are not as lucky as Xiao Hong. The symptoms of this disease were not so prominent at the beginning. Some doctors did not know enough about the disease. In addition, the penetration rate of genetic testing technology was relatively low. Many children may be misdiagnosed as a slow development like Xiao Dong.
After the symptoms, some parents thought that their children were just a simple calcium deficiency, and the best treatment window was delayed.
If you can return to 4 years ago, the first thing Xiao Dong's mother had to do is to take the child to the neurology clinic when the symptoms were just appeared.
Every small group should not be abandoned
Early diagnosis and early treatment strive for more benefits
Is it impossible to treat the child who missed the best treatment window?
In fact, as long as most children's muscles are still normal, drug treatment can relieve or even suspend the development of the disease.
Just like Xiaohong, patients who initiated the treatment before the symptoms can get a motion milestone similar to normal children. Therefore, it is critical to discover and intervene in time before the early symptoms of the disease and even the symptoms, which is the best time to cure the root cause.
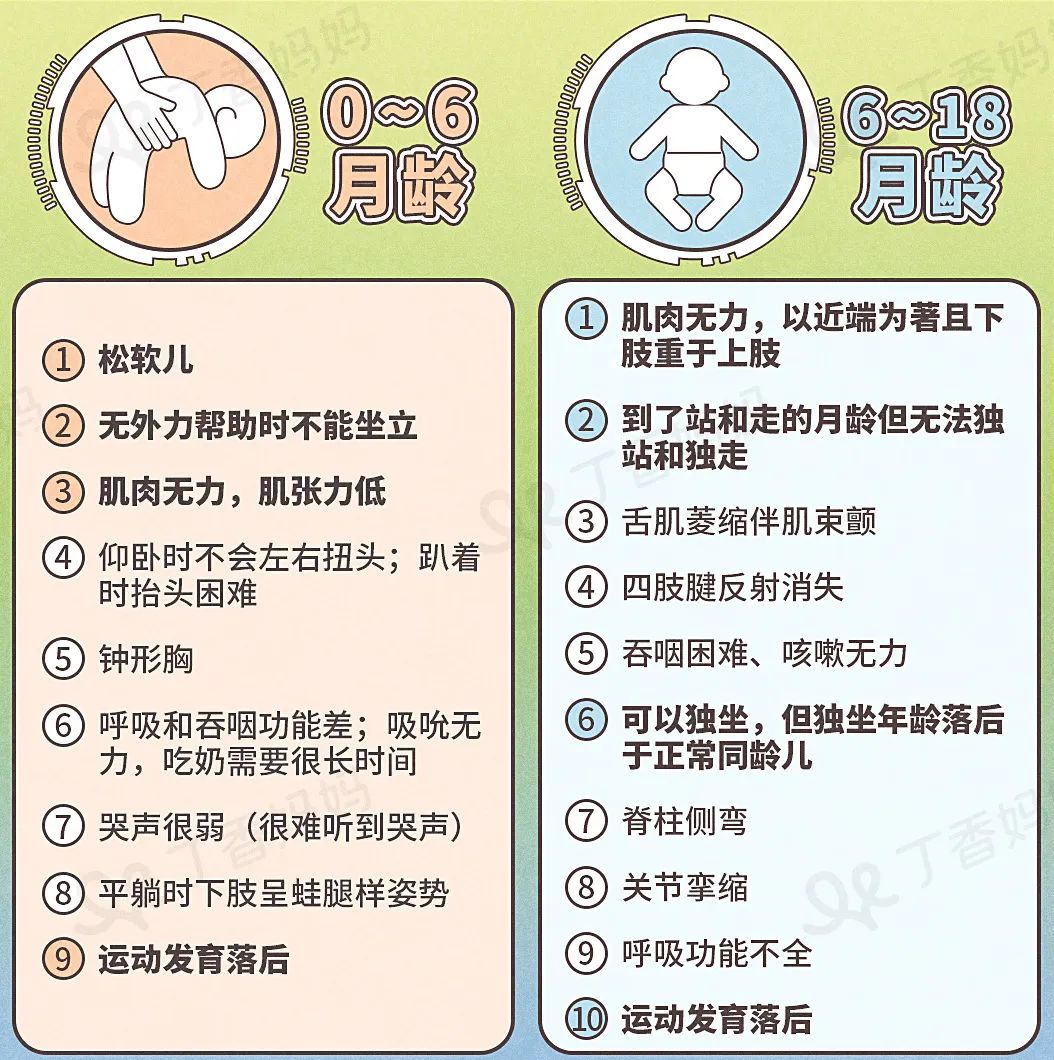
In the early stage of the disease, parents can discover signs by observing their children's milestones. If parents find that the development of the child's big movement lags behind cognition, and the mileage milestone that should have been achieved in the average month of age has not yet reached, or the following signs have appeared [10], it is recommended to seek medical treatment in time.
Figure: Early warning symptoms of SMA, baby SMA in August (in the picture, the upper color serial number represents relatively typical symptoms)
Here is a small reminder. If the child has similar symptoms, it is recommended to go to the neurology clinic, which can not only conduct a comprehensive evaluation of the child's development milestone, but also identify the diagnosis and discover problems in time, shorten the time from confirmed to intervention.
In addition to the vigilance of early symptoms, pregnant mothers (especially SMA family history) can seek genetic consultation in advance and voluntarily do SMA screening, which can detect whether the fetus occurs at the prenatal stage.
The American Society of Obstetrics and Gynecologists (ACOG) recommends that all women who are considering or pregnant should conduct SMA screening [11]. If you have no time to do pre -delivery screening, but have a SMA family history, you can also screen for screening in the newborn stage.
We do not give up every small group, but we can do a good job of prevention and early identification. There are more choices.
(The real case of Xiaodong and Xiaohong Department of the article, the name is the name of the name.)
Regarding SMA, if you still want to know more, let's take a look at the popular science of the Neuroral Medicine of Shenzhen Children's Hospital.
references
1.lunn mr, wang ch. Spinal muscular attal. Lancet. 2008 jun 21; 371 (9630): 2120-33. Doi: 10.1016/S0140-6736 (08) 60921-6. PMID: 18572081.
2. Darras bt. Spinal Muscular Atrophies. Pediatr clin north am. 2015 jun; 62 (3): 743-66. Doi: 10.1016/j.pcl.2015.03.010.
3.Sheng-Yuan Z, xiong F, Chen YJ, YAN TZ, ZENG J, Li L, ZHANG YN, Chen WQ, BAO XH, ZHANG C, xm. Molecular Characterization of SMN Copy Number Derived From Carrier Families with Sma in a Chinese Population. Eur J Hum Genet. 2010 sep; 18 (9): 978-84. Doi: 10.1038/EJHG.2010.54. , Leach Me, Finnger E. Spinal Muscular Atrophy. 2000 Feb 24 [Updated 2020 DEC 3]. In: Adam MP, Mirzaa GM, Pagon Ra, et al., Editors. GenereViews® [Internet]. Seattle (WAIVELS: Univers) of Washington, Seattle; 1993-2022. Available from: https://www.ncbi.nlm.nih.gov/Books/nbk1352/
5.Kerr DA, Nery JP, Traystman RJ, Chau BN, Hardwick JM. Survival motor neuron protein modulates neuron-specific apoptosis. Proc Natl Acad Sci U S A. 2000 Nov 21;97(24):13312-7. doi: 10.1073 /pnas.230364197. PMID: 11078511; PMCID: PMC27221.
6.Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995 Jan 13; 80 (1): 155-65. Doi: 10.1016/0092-8674 (95) 90460-3. PMID: 7813012.
7.finkel RS, Mercuri E, Darras Bt, Connolly AM, Kuntz Nl, Kirschner J, Chiriboga Ca, Saito K, Servais L, Tizzano E, TopalInius M, Montes J, GLANZMAN AM, BishoP, Gheuens s, bennett cf, school e, farwell w, de vivo dc;
- END -
The rumor of "dissolution" in response to the daily freshness: temporarily shut down Beijing -Tianjin and Shanghai Speed Dada business
Reporter Wang JunOn July 28th, the topic of Daily Fresh responses to close the 30 -minute speed appeared on Weibo hot search. Securities Daily reporter found that according to the service change n
Joint inspection of Summer City Passenger Transport Industry in Laoukou District

(Reporter: Xi Wei Wang Weijie Correspondent: Xu Jianbin Huang Chuyun)Recently, as ...